Single-Cell RNA Sequencing: A Beginner's Guide to Getting Started
Introduction
The human body contains trillions of cells and no two are identical. Within a single tumor, many distinct cell populations coexist, each with its own gene expression profile, each playing a different role in disease progression, immune evasion, or drug resistance.
For decades, scientists studied these cell populations by averaging signals across millions of cells at once — a technique known as bulk RNA-seq. The result was a blurry composite that masked the very diversity driving cancer behavior.
Single-cell RNA sequencing (scRNA-seq, also written as scRNAseq or single cell RNA sequencing) has changed that. By profiling the transcriptome of each individual cell, this technology delivers gene expression data at single-cell resolution, revealing rare subpopulations, transitional cell states, and cellular hierarchies that bulk methods cannot detect.
When combined with single-nuclei RNA sequencing (snRNA-seq) — which extracts nuclei rather than whole cells to enable profiling of frozen or fixed tissues — researchers now have a comprehensive toolkit for mapping the full cellular landscape of any tissue or tumor sample.
This guide provides an evidence-based overview of single-cell transcriptomics, its role in cancer research, its application in patient-derived xenograft (PDX) models, and the end-to-end single-cell PDX workflow offered by Admera Health.
What Is Single-Cell RNA Sequencing (scRNA-seq)?
Single-cell RNA sequencing is a next-generation sequencing technique that measures the gene expression profile of individual cells within a mixed population. Unlike bulk RNA-seq, which averages the signal from thousands to millions of cells, scRNA-seq isolates each individual cell into its own reaction vessel, barcodes its RNA molecules, and sequences them independently. The result is a high-dimensional dataset where active researchers can see thousands of genes expressed simultaneously — at the level of a single cell.
A closely related approach, single-nuclei RNA sequencing (snRNA-seq), follows the same logic but works with isolated nuclei instead of intact cells. This makes it especially valuable for tissues that are difficult to dissociate — frozen biobank samples, solid tumors, or archived PDX tissue where intact cell isolation would compromise sequencing data quality.
Key Differences between ScRNA-seq and SnRNA-seq
ScRNA-seq works from fresh or lightly fixed dissociated cells, while snRNA-seq works from nuclei extracted from frozen or FFPE tissue. Both generate a single-cell transcriptome, but the right choice depends on sample type and availability.
Together, scRNA-seq and snRNA-seq allow researchers to:
Identify and classify distinct cell types and subtypes within a complex tissue
Detect rare cell populations representing fewer than 1% of total cells
Track cell-state transitions, differentiation trajectories, and lineage relationships
Map interactions between tumor cells, immune cells, and stromal cells
Pinpoint genes and pathways driving drug resistance or immune evasion
Its high specificity and sensitivity make single-cell sequencing a powerful approach for clinical research and molecular diagnostics — and nowhere has its impact been felt more acutely than in cancer research.
Single-Cell Transcriptomics in Cancer Research
Cancer is not a disease of one cell type behaving badly. It is an ecosystem of distinct cell populations interacting dynamically and single-cell RNA sequencing has given researchers the tools to map that ecosystem for the first time. It reveals the subpopulations that drive resistance, evade immunity, and ultimately determine whether a patient responds to treatment.
Mapping Tumor Heterogeneity
Tumors are ecosystems composed of malignant cells at various stages of differentiation, immune cells, fibroblasts, endothelial cells, and more — all interacting dynamically. These seq methods have illuminated tumor biology, immune escape mechanisms, and treatment resistance in ways that conventional bulk-tissue methods could not achieve.
Bulk RNA-seq, because it averages signals across mixed cell populations, often fails to resolve clinically relevant rare cellular subsets. This limitation is particularly costly in cancer research: a small population of drug-tolerant cells can drive relapse even when the bulk of the tumor appears treatment-sensitive. To understand those individual cells, you need single-cell resolution.
Head and Neck Cancer: A Case Study
Head and neck squamous cell carcinoma (HNSCC) causes nearly 400,000 deaths annually and remains one of the most difficult cancers to treat with immunotherapy. A landmark study by McCord et al. (2026), published in Science Immunology, illustrates how single cell RNA sequencing is redefining our understanding of why T-cell presence does not reliably translate into therapeutic response.
By applying scRNA-seq and TCR sequencing to paired blood and tumor samples from 27 HNSCC patients, the team defined the full transcriptional landscape of tumor-infiltrating T-cells at single-cell resolution. They distinguished genuinely tumor-reactive clones from immunological bystanders — a distinction that bulk sequencing simply cannot make.
The TCR sequences identified through scRNA-seq were then used to design patient-specific probes for spatial transcriptomics, enabling physical mapping of each T-cell clone within intact tumor tissue.
This revealed that intratumoral T-cell states differed markedly from those in peripheral blood, varied substantially across patients, and were shaped by both antigen specificity and location within the tumor microenvironment. Without scRNA-seq as the foundational layer, that high-resolution spatial mapping could not have been executed.
The Multi-Omics Frontier
The field is rapidly moving beyond transcriptomics alone. Single-cell multi-omics approaches integrate genomic, epigenomic, proteomic, and spatial information from the same sample — enabling simultaneous profiling of what a cell is expressing, which regions of its genome are accessible, and where it sits within tissue architecture. This level of biological detail is increasingly guiding the design of precision immunotherapy and targeted therapy strategies across cancer types.
Admera Health's Single-Cell Workflow
Admera Health provides an integrated single-cell RNA sequencing service that combines wet-lab infrastructure with a dedicated bioinformatics team — giving researchers a single point of accountability from sample receipt to final report.
Every stage of the workflow is designed to protect sample integrity and analytical rigor, from the moment a sample arrives through to bioinformatics delivery.
Sample Requirements and Accepted Input Types
Admera's workflow accepts both cell/nuclei and tissue submissions across a range of preservation states. Accepted inputs include fresh and cryopreserved cells or nuclei (recommended >1M cells per sample in duplicate; minimum 200K), fixed cells or nuclei, fresh tissue (100 mg), flash-frozen tissue (50 mg), FFPE tissue (minimum 2 sections), and PFA-fixed tissue. View the full sample submission guide here.
Cell viability at receipt determines the processing route. Samples above 90% viability are ideal; 70–90% is acceptable; 50–70% requires dead cell removal before library preparation; below 50% viability fails QC.

This tiered approach ensures that every sample entering the pipeline meets the quality threshold required for reliable sequencing data. Admera recommends a mock shipment in advance to confirm cells maintain acceptable viability under your specific transit conditions.
Library Preparation and Sequencing
Admera's library preparation workflow is optimized for consistency and reproducibility across sample types.
Step 1 — Sample Preparation and Quality Check Tissue is dissociated into a single-cell or single-nucleus suspension, or tissue sections are mounted onto spatially barcoded slides or chips. Concentration, integrity, and viability are confirmed before moving forward.
Step 2 — Molecular Barcoding Each cell, nucleus, or spatial position is assigned a unique molecular barcode linking every downstream sequencing read to its origin — achieved through microfluidic GEM encapsulation, combinatorial split-pool barcoding, or in situ capture on spatially arrayed oligonucleotides.
Step 3 — Library Construction Barcoded RNA undergoes reverse transcription into cDNA, amplification, and conversion into sequencing-ready libraries. Certain sequencing platforms generate parallel libraries from the same barcoded material — for example, paired gene expression and immune receptor libraries for V(D)J profiling. All libraries undergo QC by Bioanalyzer profiling and quantitative PCR before sequencing.
Step 4 — Sequencing Libraries are sequenced to a target read depth appropriate for the chosen assay. Short-read sequencing suits gene expression quantification and chromatin accessibility profiling. Long-read sequencing enables full-length transcript reconstruction and isoform resolution. The approach is confirmed during project scoping with the Admera team.
Bioinformatics Analysis Pipeline
Raw sequencing data are processed through Admera's validated single-cell bioinformatics pipeline. Analysis approach differs by assay and sequencing technology as noted.
Step 1 — Alignment and Quantification Reads are demultiplexed, quality-trimmed, and aligned to the appropriate reference genome using Cell Ranger, STARsolo, Space Ranger, or pbmm2 depending on platform. Multi-species alignments are supported where required. For long-read data generated on the PacBio platform, reads are processed using SMRT Link Read Segmentation and Single-cell Iso-Seq workflow process and aligned with pbmm2, enabling full-length transcript quantification and isoform-level resolution.
Step 2 — Quality Control and Filtering Cells are filtered by detected genes, total UMI counts, and mitochondrial gene percentage. Doublets are identified and removed. For long-read seq data, additional QC steps assess read length distribution and full-length transcript rate.
Step 3 — Dimensionality Reduction and Clustering Filtered cells are normalized, scaled, and projected into low-dimensional space using PCA and UMAP. Unsupervised clustering identifies distinct cell populations based on transcriptional similarity. For multi-omic data, clustering uses weighted nearest neighbor analysis across both chromatin accessibility and gene expression.
Step 4 — Cell Type Annotation Clusters are annotated using curated marker gene sets, distinguishing malignant subpopulations, stromal cells, tumor-infiltrating immune cells, and endothelial populations. For spatial datasets, each cell type is mapped back onto tissue coordinates to contextualize populations within their anatomical microenvironment.
Step 5 — Differential Expression and Pathway Analysis Differentially expressed genes between clusters or experimental conditions are identified using statistical frameworks appropriate for single-cell count data. Differential expression analysis is one of the most informative outputs of any scRNA-seq experiment — mapping gene expression changes to processes like immune signaling, cell cycle regulation, and drug resistance pathways.
Step 6 — Deliverables and Report Admera provides a comprehensive analysis report including UMAP visualizations, cluster annotations, cell type proportion summaries, differentially expressed gene lists, pathway enrichment results, and QC metrics. Isoform classification summaries are included for long-read experiments. Reports are structured to meet the standards of peer-reviewed publication and regulatory submission — so your data is ready to use the moment it arrives.
Choosing the Right Sequencing Method:
Not sure which sequencing method fits your experiment? Here is a straightforward decision tree to guide your choice when analyzing your single-cell data:
Fresh, dissociated cells with >70% viability → scRNA-seq
Frozen, FFPE, or difficult-to-dissociate sample → snRNA-seq
Need spatial tissue context → Add spatial transcriptomics
Need isoform-level resolution → Long-read single-cell sequencing
Comparing multiple treatment conditions → Multiplexed sequencing
When in doubt, Admera's project management team can help you navigate the decision based on your sample type, experimental goals, and budget.
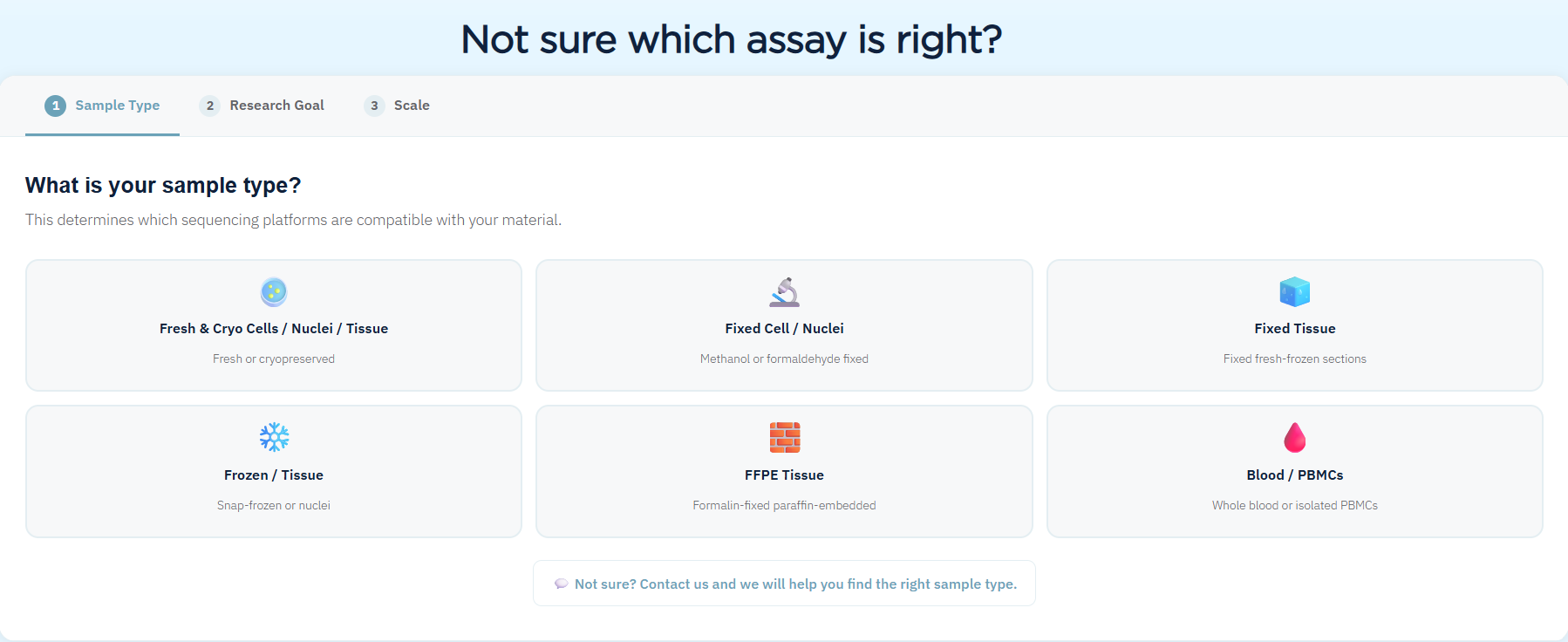
Select the right single-cell sequencing platform based on sample type including fresh cells, frozen tissue, FFPE, and fixed samples for scRNA-seq or snRNA-seq.
The Long-Read Frontier: Expanding What scRNA-seq Can Resolve
Standard short-read sequencing platforms read 150 to 300 base pairs per fragment — a window too narrow to capture full RNA isoforms, alternative splicing events, or structural variants. Researchers asking not just which genes are expressed in a cell, but how those genes are assembled into functional transcripts, find that short-read seq data alone cannot provide the full answer.
Long-read sequencing platforms are closing this gap. PacBio has achieved 99.9% sequencing accuracy, generating reads of 1–3 kb for most cDNA and up to 15 kb for concatenated libraries. This enables full-length RNA isoform identification and resolution of complex alternative splicing patterns at single-cell resolution — something short-read approaches cannot replicate regardless of sequencing depth.
For cancer research, this opens an additional dimension of expression analysis. Long-read scRNA-seq can characterize structural variants, gene fusions, and isoform-level expression changes within individual cells, resolving biological signals that would otherwise be lost in both bulk sequencing and standard short-read scRNA-seq.
Conclusion
Single-cell RNA sequencing has fundamentally changed what is possible in cancer research. By resolving tumor biology at the level of individual cells, scRNA-seq and snRNA-seq enable scientists to identify therapeutic targets, map resistance mechanisms, and characterize the tumor microenvironment with a precision that bulk sequencing simply cannot match.
Applied to patient-derived xenograft models, single-cell transcriptomics unlocks an additional dimension: the ability to cleanly separate human tumor biology from murine host responses, interrogate PDX heterogeneity at single-cell resolution, and track how tumor cell populations evolve across treatment conditions.
For research teams working with PDX models, the integration of single-cell RNA-seq is no longer a future aspiration — it is an active, accessible capability.
Admera Health's end-to-end single-cell PDX workflow, spanning sample receipt through bioinformatics delivery, is designed to make that capability accessible without compromising analytical rigor.
